Thalassemia
introduction
The Thalassemia is a hereditary conditional Disease of the red blood cells. Here is that hemoglobin, an iron-containing protein complex that is responsible for the oxygen-binding ability of the red blood cells. It will not formed in sufficient quantity, or increased reducedso that a Hemoglobin deficiency results.
Depending on the severity of the thalassemia, it is a serious clinical picture that, if left untreated, can be fatal in early childhood. Thalassemia is particularly widespread in the Mediterranean region. This is where the name comes from, because thalassemia means something like "Mediterranean anemia". Mostly people in former Malaria areas affected, for example in Malta, Cyprus, Greece and Sardinia. This is because the mild form of thalassemia has an evolutionary advantage in malaria diseases. The genetic defects in the red blood cells prevent the malaria pathogens from multiplying in the red blood cells. As a result, humans had a survival advantage and thalassemia was able to establish itself further in the course of evolution.
causes

The thalassemia can turn into a alphaShape and one beta-Form can be divided. Both forms of the disease are specific Genetic defects underlying.
In the Alpha thalassemia the gene mutation is on Chromosome 16, the far more common Beta thalassemia the defect is on Chromosome 11.
The alpha-Thalassemia is generally characterized by a milder course.
The beta-Thalassemia can be further divided into a minor and a major sub-variant.
Which form is present depends on the inheritance of the disease. The disease will autosomal recessive inherited, that means only patient who two diseased features (Alleles) inherit from their parents, from the severe form of thalassemia - thalassemia major - get sick. This form is also called homozygous Variant designated. If the patient only inherits one sick allele and also has a healthy allele, one also speaks of one heterozygous Variant, the thalassemia minor.
Symptoms

The symptoms of thalassemia differ depending on the type of thalassemia.
Patients with alpha thalassemia tend to have fewer symptoms overall. However, alpha thalassemia can also be present in different degrees of severity. In the most severe form, the unborn child can die in the womb.
General symptoms of thalassemia come from anemia (anemia) conditions. These include exhaustion, tiredness, dizziness, poor performance, racing heart, headache and shortness of breath during physical exertion. Severe forms of the course can be caused by jaundice (Jaundice) through the breakdown of red blood cells and a greatly enlarged spleen.
Beta-thalassemia in its minor form is usually not manifested by clinical symptoms, as the healthy gene outweighs the diseased gene in its function. In some cases, these patients have a slightly enlarged spleen. Otherwise there are usually no complaints.
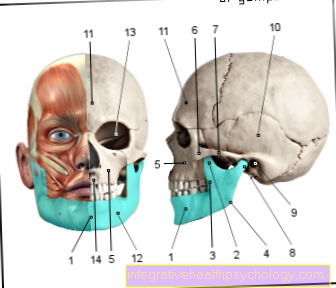
The major form of beta thalassemia (also: Cooley anemia) behaves significantly differently. These patients only have the gene in its diseased form and thus develop severe general symptoms. Children who are born with this disease are noticed shortly after birth with a massive enlargement of the liver and spleen. They suffer from growth retardation, severe organ damage and malformations of the bone. Since the bone marrow makes the blood, but the disease does not function properly, the bone marrow is stimulated to overproduce red blood cells. This is ultimately expressed by a thickening, especially of the cheek and skull bones, resulting in a wider eye relief and enlarged cheekbones. As with alpha thalassemia, the symptoms are also caused by anemia (fatigue, tiredness, dizziness, poor performance, racing heart, headache and shortness of breath during physical exertion). Jaundice (Jaundice) due to the increased breakdown of red blood cells.
diagnosis

The suspicion of thalassemia in a sick child can often be caused by the Family history come up. Has the child sick relatives, it is likely that it also inherited the defective gene.
By examining the blood in the laboratory and under that microscope the suspicion can be confirmed. It is typical that the Iron value in the blood in thalassemia normal is. This distinguishes the disease from the clinically similarly impressive one Iron deficiency anemia where the iron value is too low. The red blood cells in thalassemia are im microscope often as so-called Target cells to see (Target-like coloration of the red blood cells).
Other clinical symptoms of the patient, such as a enlarged spleen and Symptoms of anemia can also confirm the diagnosis. Last but not least, a molecular genetic examination can be undertaken to specifically search for the genetic defect. This can be relevant for couples who want to have children if they want to determine the risk of having a sick child.
therapy

The minor-Form of beta-Thalassemia, as well as the mild forms of alpha-Thalassemia usually do not require any medical therapy at all.
The severe major-Thalassemia must, however treated compulsorily otherwise it leads to the death of the patient within a short time. The severe form of beta thalassemia is usually treated with regular administration of Blood transfusions treated. The patients receive several blood units 2-4 weeks apart. This makes the patient healthy and functional blood and suppresses the pathological blood formation in your own body. Side effects of thalassemia, such as Growth retardation and lack of skeletal maturity, can thereby largely prevented become.
The problem with the continuous administration of blood transfusions goes hand in hand is that Overloading the body with iron. The blood reserves become more iron introduced into the body than can be excreted. The excess iron is deposited in the internal organs and in the long run can lead to serious organ malfunctions there. Therefore, an increased iron excretion has to be achieved therapeutically. This is achieved by means of so-called Chelating agents (Substances that form complexes with excess iron and lead to its excretion). These drugs can be used as either infusion via the vein or as tablet administered. Therapy with blood transfusions and chelating agents must lifelong respectively.
The spleen is nowadays reluctant to remove because it is important for the immune system.
The only way to cure the disease at the moment is Stem cell transplant. It will Bone marrow of the patient through a chemotherapy destroyed and then rebuilt by healthy stem cells from a donor. However, since stem cell transplantation is also associated with many risks, this procedure must be weighed individually for each patient and checked to see if it makes sense.
forecast
The prognosis of thalassemia is strongly dated Degree of expression dependent on the disease.
Patient with mild forms can usually be in normal life lead without major restrictions.
In the severe It depends on the effectiveness of the therapy and on occurring Complications on. In a conversation with the attending physician, the prognostic prospects of the disease in the individual patient can be discussed.
prophylaxis
The thalassemia cannot be prevented prophylactically because it is a hereditary Gene mutation. However, people from affected families can choose to have children genetic advice and have your own risk of having a sick child determined.