What is Alpha-Galactosidase?
Alpha-galactosidase is an enzyme that belongs to the group of hydrolases. Another name for the enzyme is ceramide trihexosidase.
The enzyme occurs in all human cells and cleaves the alpha-D-glycosidic bond.
An alpha-glycosidic bond is a bond between a carbohydrate such as galactose and an alcohol group, for example.
More precisely, the enzyme alpha-galactosidase occurs in the lysosome of cells and breaks down glycosphingolipids and glycoproteins into their individual components.
If the enzyme is defective due to a genetic error, Fabry disease can develop.

Task, function and effect of alpha-galactosidase
Alpha-galactosidase is an enzyme and its job is to help break down special fats. These fats can be absorbed into the body through food.
They occur in virtually all cells in the body. The breakdown of fats by alpha-galactosidase prevents pathological reproduction within the cells. The functioning of the enzyme corresponds to that of a classic hydrolysis. During hydrolysis, the chemical bond (alpha-D-glycosidic bond) between the individual components of a molecule is split with the incorporation of a molecule of water, resulting in two products.
Due to the typical properties of the enzyme, the splitting reaction of the fats is significantly accelerated. As a result, more fats can be broken down per unit of time.
The enzyme alpha-galactosidase emerges from this reaction like all enzymes unchanged and in the same amount. The catalyzed reaction of alpha-galactosidase has the effect of reducing the total amount of glycosphingolipids in the body.
Glycosphingolipids are the names of the fats that are broken down by alpha-galactosidase.
Overall, this prevents the death of the body cells due to the accumulation of glycosphingolipids in the cells. This means that the clinical picture of Fabry's disease does not occur.
Where is it made?
Precursors of the enzyme alpha-galactosidase are synthesized by the ribosomes of the cells. These release a chain of amino acids, from which the finished enzyme is later created, into the endoplasmic reticulum.
Here, maturation takes place to the finished enzyme. From there, the amino acid chains are transported to the Golgi apparatus through special vesicles. From here the enzymes can either be transported through vesicles into the lysosomes or to the cell surface and released into the space between the cells.
Other cells can now take up the enzymes in a receptor-mediated manner.
Alpha-galactosidase decreased
A deficiency in the enzyme alpha-galactosidase results in a reduced breakdown of fats (glycosphingolipids) in the body's cells.
This leads to an accumulation of fats in the lysosomes of the cells.
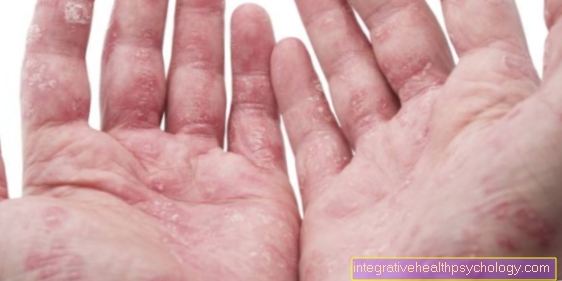
This accumulation is poorly tolerated by most cells and has harmful effects on them. As a result, programmed cell death often occurs. This can manifest itself in various symptoms such as changes in the skin and blood vessels or pain in the hands and feet.
Fabry disease
Fabry disease is a disease caused by a deficiency in alpha-galactosidase. The cause of the deficiency is usually hereditary and very rare.
The disease is often only noticed when several symptoms appear in late childhood.
There are punctiform skin changes, pain in the hands and feet, a change in temperature sensation, hearing loss, changes in the eyes and protein in the urine.
Changes in the blood vessels cause the kidneys in particular to be threatened by a kidney infarction or renal insufficiency.
In addition, the heart threatens a heart attack. A stroke also often occurs. Therapy consists in the early substitution of the enzyme with tablets. Dialysis or a kidney transplant may be necessary as the disease progresses.
For detailed information on this topic, see: Fabry disease
Can you substitute them with tablets?
If there is a deficiency in the enzyme alpha-galactosidase, this can be contained very well by taking tablets, so that hardly any changes are noticeable.
The tablets contain artificially produced enzymes, the function of which corresponds exactly to the body's own enzymes.
Substitution with tablets is the generally used therapy in the case of a known deficiency in alpha-galactosidase. For this reason, the therapy is also known as enzyme replacement therapy.
Because the cells of the body have special surface receptors that recognize the enzyme, the cells can endocytose the enzyme into their plasma. After absorption, the enzymes develop their effect within the cell, which lowers the amount of glycosphingolipids.
Alpha-galactosidase increased
An increased amount of alpha-galactosidase does not play a role in today's medicine. No negative effects of a large amount of the enzyme alpha-galactosidase on humans have been described.
An increased amount of alpha-galactosidase usually only occurs if too large an amount has been consumed in the case of substitution with tablets.








.jpg)